Introduction
As discussed in previous blog posts, the scale of modern-day NGS instruments has opened a world of possibilities for sequencing hundreds, and even thousands, of samples simultaneously. Multiplexing is now a core capability that drives the utilization of sequencing in a wide variety of research and production-scale assays. Past blogs have outlined how multiplexing works at the level of library preparation, but there is another key aspect to NGS—that different samples can be distinguished from each other once they are pooled together on the same sequencing run.
“Indexing” or “barcoding” is a broad term that describes how additional sequences are added to libraries and read on sequencers to make this happen. Barcoding is widely used in genomics research and invariably involves attaching short nucleotide sequences, called sequence barcodes or indexes, to DNA fragments during library preparation and then reading and deconvoluting this information on sequencing instruments.
Barcoding serves to identify and sort DNA fragments, enabling researchers to analyze more samples during a single run. By increasing the efficiency, cost-effectiveness, and throughput of sequencing experiments, barcoding helps advance the possibilities of NGS.1
There are a wide range of uses for different types of NGS barcodes in various sequencing applications. In many cases, the type or style of barcode will be influenced by the technical requirements of different assays. In this article, we review how to select the best barcoding method, depending on your application, sample type, and budget. We also discuss how different library preparation workflows enabled by seqWell technologies implement different styles of indexing for various applications.
Barcode Anatomy: i5 and i7 Index Sequences
Illumina-based sequencing is by far the most widely utilized NGS platform in life sciences, and therefore Illumina-style index design is a nearly universal model for how indexed sequencing is performed. Illumina library barcoding involves designing and adding DNA sequences at specific positions on either end of each DNA fragment during library preparation. These index positions are called i5 and i7, matching the terminal P5 and P7 sequences used for clonal amplification on the flow cell.
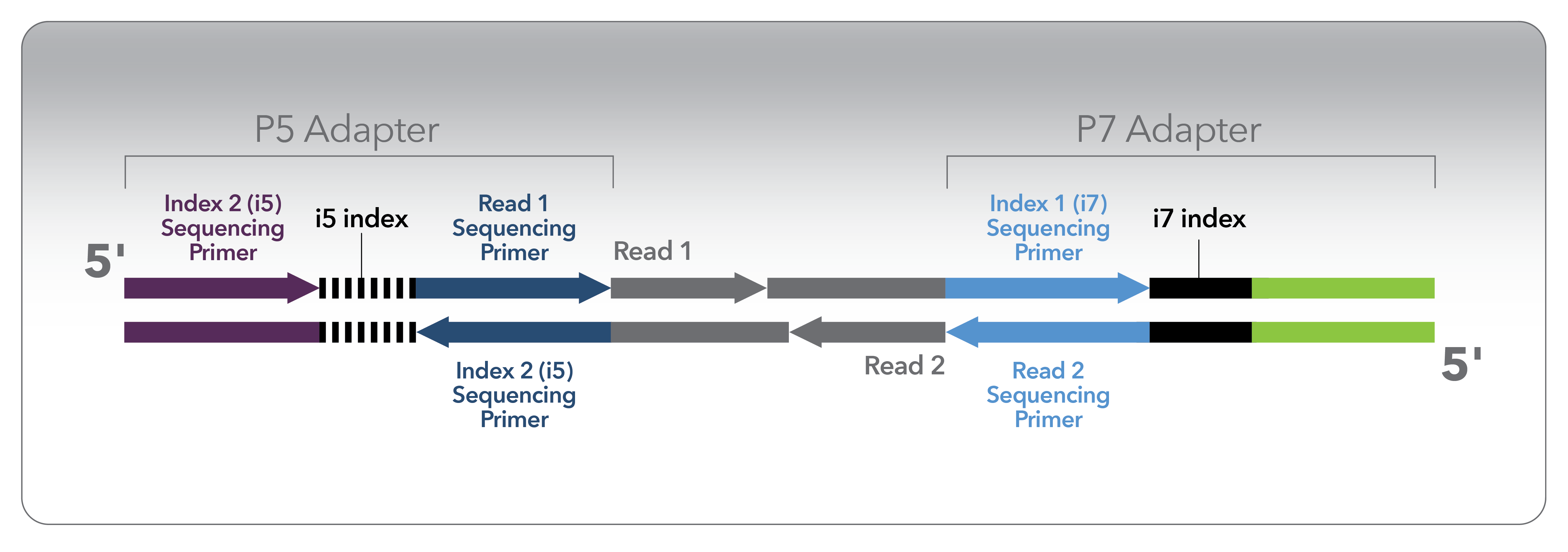
It is also possible to index libraries with barcodes in positions other than i5 and i7, such as inline barcodes that are read as part of the sequencing insert itself (i.e., R1 or R2). However, sample-level multiplexing is more commonly performed via barcodes at the i5 and i7 positions. i5 and i7 indexes are typically 8 or 10 nucleotides long. In some cases, the indexes can be longer than 10 nucleotides, especially when the barcode contains a unique molecular identifier (UMI) as well as a sample index.
Adding Barcodes to Libraries
Barcoding that uses i7 and i5 indexes occurs during library preparation, after DNA is extracted from the samples and processed to create a library. At this point, barcodes can be added using one of the following methods:
- Using PCR primers with different combinations of i5 and i7 index sequences to amplify libraries that have been adapted with PCR-compatible adapter sequences.
- Using ligatable adapters that have different combinations of i5 and i7 index sequences.
- Using transposase-based methods to barcode with indexed transposase tagging reagents.
In RNA sequencing, indexes may be added while converting RNA to readable DNA molecules, such as barcoding of molecules during reverse transcription.2 This workflow is commonly used when UMI indexes are added to label each molecule in a sample with a unique sequence before PCR application begins.
Combinatorial and Unique Dual Indexing
Dual indexing refers to a family of barcoding methods that uses combinations of i5 and i7 indexes to identify individual samples within a pool of libraries that are sequenced together.3 There are two types of dual indexing: combinatorial dual indexing (CDI) and unique dual indexing (UDI).4
CDI uses each i5 and i7 index multiple times but only uses each combination of indexes once. Therefore, the index combination used to tag each sample is unique, but the individual indexes are not. For example, 96 unique index combinations can be created from eight i5 indexes and twelve i7 indexes.
In contrast, UDI uses each i5 and i7 index only once so each combination and index are both unique. For example, creating 96 unique index combinations requires 96 unique i7 and 96 unique i5 indexes.
When are unique dual indexes required? Some applications are particularly susceptible to any amount of contamination—or crosstalk— between index sequences. For example, analysis of sensitive counting applications may be affected if even a small number of reads from one sample are contaminated.
Index hopping is a form of contamination that can affect experiments that use barcoding, especially on systems that use patterned flow cell technology.5,6 Index hopping happens when samples are incorrectly assigned to the wrong index during demultiplexing. Typically, it occurs in less than 1% of total reads.
UDI barcoding mitigates the impact of index hopping because both the i5 and i7 indexes must match an expected combination. Any reads with unexpected combinations are removed from downstream analysis. However, this results in a higher rate of discarded data compared to CDI.
When libraries are amplified as a pool, such as in hybrid capture applications, there is also a low-level risk of PCR-induced chimeras, which can also cause i5 and i7 index sequences to be misincorporated and read incorrectly during sequencing.7 For these applications, UDI indexing is also strongly preferred.
Unique Molecular Identifiers
UMIs are random or semi-random index sequences that are unique to each molecule in a sample library.8 UMIs are made by integrating an adapter sequence that has been synthesized to include nucleotides that are randomized at specific positions (sometimes i5 or i7, or in-line to the sequencing insert). This process usually occurs during ligation.
After sequencing, UMIs are processed by bioinformatic tools that allow for otherwise identical-looking sequenced molecules (i.e., part of a transcript) to be distinguished as unique if they have a different UMI sequence.
Demultiplexing
After pooled libraries are sequenced, a demultiplexing algorithm uses these unique combinations of indexes or UMIs to sort the sequenced reads according to which sample they came from. In other words, demultiplexing is the splitting of the raw sequencing data into separate files using sample-specific barcodes.9
Effective demultiplexing is key to accurately sequencing multiple libraries at once. This step occurs before final data analysis.
What Makes A Good NGS Barcode?
Commercially available NGS library preparation kits come with various index design features. Selecting the appropriate index sequence design and protocol for your NGS experiment is crucial for maintaining efficient and accurate sequencing runs. It’s important to ensure that each barcode is reliably matched to the correct sample—even if the sequencing system makes an error.
To facilitate accurate demultiplexing and prevent the sequencing data of one sample from “contaminating” others on the same run, barcodes are designed with features like minimum Hamming distance—a metric that describes how different the barcodes are from each other.10 Commonly, barcode sets are designed with a minimum Hamming distance of at least 3, meaning that any two barcodes must differ in at least 3 positions, so that any particular barcode can tolerate at least one error.
Barcodes are designed and used in various ways depending on the specific experiment and sequencing platform used.
Choosing a Barcode Strategy That’s Best for You
There are several important factors to consider when selecting different designs or combinations of index sequences. The required number of samples, the potential for contamination or crosstalk, and workflow speed and time all influence the optimal choice of index configuration for different projects. For this reason, seqWell products are designed with a range of different indexing requirements and configurations in mind.
When a large number of samples need to be sequenced cost-effectively, CDI enables more i5 and i7 index combinations to be created within a tighter budget. For this reason, CDI supports experiments that require high-scale multiplexing of thousands of samples per run, such as synthetic construct sequencing.
For multiplexing up to 2,308 samples using CDI, seqWell offers a fleet of plexWell™ library preparation kits. Available in many configurations, plexWell supports applications like whole genome sequencing, scRNA-seq, and metagomics/microbiome sequencing, in addition to synthetic construct sequencing.
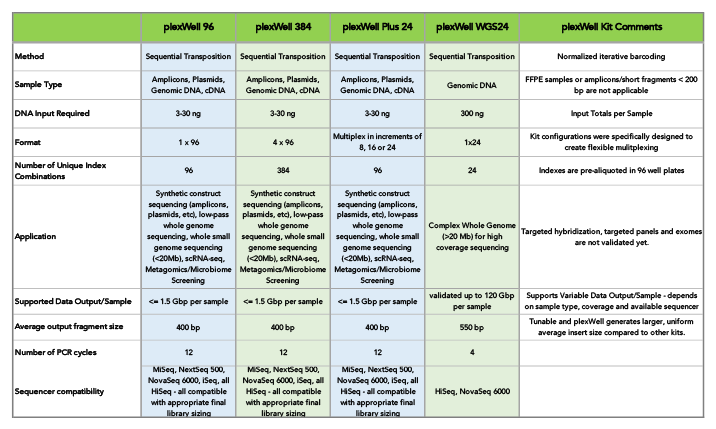
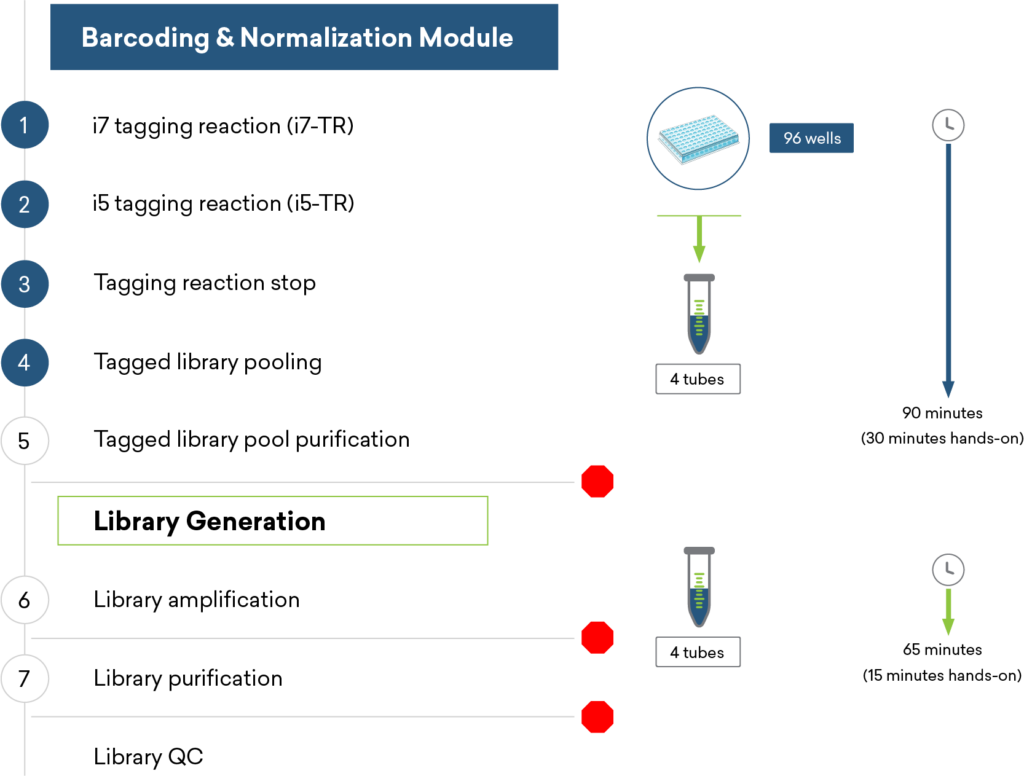
For additional efficiency in multiplexed plasmid and amplicon sequencing, the ExpressPlex™ Library Prep Kit enables one-step library prep with CDI barcoding in just 90 minutes, while simultaneously auto-normalizing library read-count and insert size.
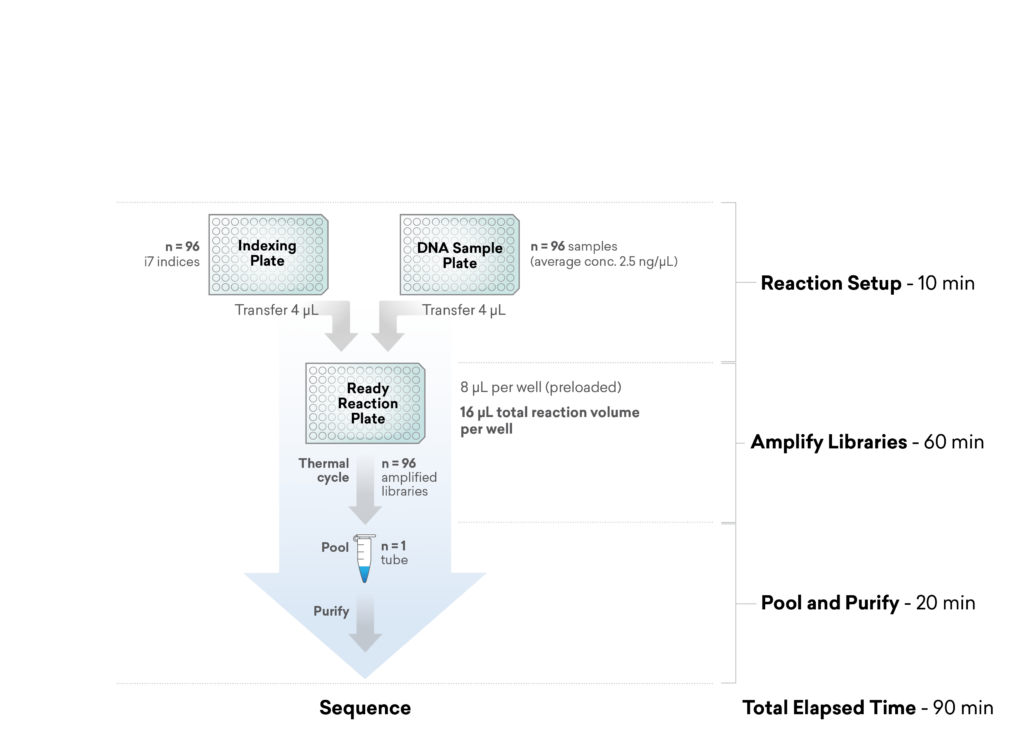
The rate of index hopping will vary between library prep kits and must be balanced against considerations of labor time and cost.
The purePlex™ Library Prep Kit reduces workflow burden through auto-normalization of UDI libraries that permit pooling of samples immediately after sequential transposase mediated tagging steps. purePlex offers reduced QC and labor costs compared to traditional UDI workflows.
An Eye Toward the Future
As sequencing technologies continue to expand, so will the need for better tools to use these platforms more effectively. Likewise, the creation of kits and tools that afford a wide range of different multiplexing configurations and performance features will continue to be an important area.
One of our primary goals at seqWell is to make products that make sequencing workflows more scalable and efficient across a wide range of multiplexing applications. A core foundation for this work has been creating purpose-built NGS indexing products like plexWell and purePlex, and further building on this power with new products such as Tagify™ and ExpressPlex.
The future of sequencing will depend on more barcodes, and seqWell will be there!
References
- https://www.nature.com/articles/s41598-017-12825-2
- https://www.researchgate.net/figure/Two-methods-to-add-barcode-in-a-single-cell-a-cDNA-is-reverse-transcribed-and-amplified_fig3_327632896
- https://www.ogc.ox.ac.uk/wp-content/uploads/2017/09/indexed-sequencing-overview-guide-15057455-03.pdf
- https://seqwell.com/when-unique-dual-indexes-matter/
- https://www.illumina.com/techniques/sequencing/ngs-library-prep/multiplexing/index-hopping
- https://www.illumina.com/science/technology/next-generation-sequencing/sequencing-technology/patterned-flow-cells.html
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC92662/
- https://www.lexogen.com/rna-lexicon-what-are-unique-molecular-identifiers-umis-and-why-do-we-need-them/
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8287537/
- https://tools.epigenetic.ru/ts-doc/ion-docs/GUID-BA1FED7A-5809-46DD-8E0E-B1DEDF596D23.html