Introduction
This article is the second in our series that highlights seqWell’s multimedia content offerings. The following is a transcript of our recent webinar: Assessing CRISPR On-Target Editing and Structural Changes with UDiTaS™ using Tagify™ Reagents1. This work was also highlighted in a recent poster presentation given at the Advances in Genome Biology and Technology (AGBT 2023) meeting in Hollywood, FL, on Feb 6-9, 2023, which can be viewed here.
Jack Leonard, PhD, seqWell’s Co-Founder and Chief Technology Officer, opened the presentation with background on seqWell’s transposase-based library prep technology. Then Georgia Giannoukos, Director of Next Generation Sequencing at Editas Medicine, highlighted how her organization has leveraged seqWell’s technology to ensure consistency and reproducibility2.
A Toolbox of Transposase-Based Technology
Jack Leonard: Before I hand off the presentation to Georgia, I’d like to give a brief introduction to seqWell’s library preparation technologies. And the enzyme at the heart of seqWell’s library preparation biochemistry is the Tn5 transposase protein. Tn5 transposase is particularly versatile for scaling and simplifying NGS workflows because it enables simultaneous fragmentation of the target DNA and attachment of synthetic adapters onto library fragments in a single step.
Tagify is the name of our adapter-loaded Tn5 transposase reagents, which are available as standalone tagging reagents. We will be focused on Tagify UMI tagging reagents today, but it’s worth mentioning that this is only one of the tools in seqWell’s toolbox.
Tagify adapters are full-length and include the p5 primer, eight base i5 barcode, followed by a randomized 10 base sequence or UMI. And finally, at the 3′ terminus there is a 19 bp mosaic end sequence, which serves as the binding sequence for Tn5 transposase.
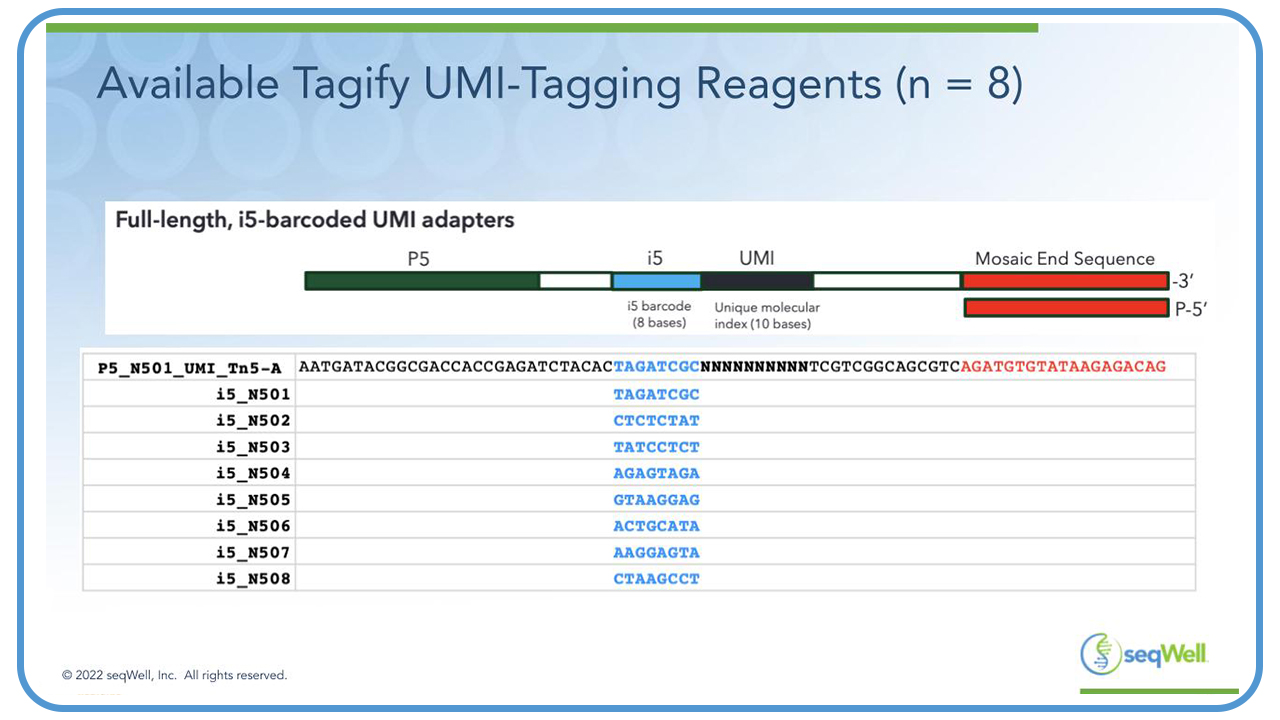
The bottom panel on this slide shows the sequence of the adapters on the eight different Tagify UMI tagging reagents that are available from seqWell, and these can be distinguished from each other by their eight base i5 barcodes. These are the same designs as described by Georgia in her UDiTaS publication3.
So, the Tagify UMI tagging reagents insert semi-randomly into genomic DNA to create nested series of library fragments. Now that these Tagify UMI tagging reagents are available from seqWell, we believe it will enable others to use this powerful UDiTaS method that Georgia is about to describe to you.
Learning Objectives
Georgia Giannoukos: Thank you, Jack. Today I’ll be talking about the Tagify reagents and how we use them for our UDiTaS process to assess CRISPR on-target editing and structural changes.
First, I’ll give a quick overview of the CRISPR Cas9 editing system. Then, I’ll go over our UDiTaS method and how we use it to measure structural changes, specifically measuring large deletions and inversions at the CEP290 editing site. We can measure inter-chromosomal translocations, and we’ve developed accuracy standards for structural changes.
Finally, I’ll talk about Tagify, which we use as a custom UDiTaS reagent in our process. I’ll show you data comparing different batches showing reproducibility. And finally, I’ll talk about how we’re scaling our reaction from 96 to 384 well reactions.
CRISPR Cas9 Editing System
So, in the CRISPR system, RNAs are the targeting component of the Cas9 enzyme. This is a complex of the Cas9 nuclease with the guide RNA over the genomic DNA target site for cutting.
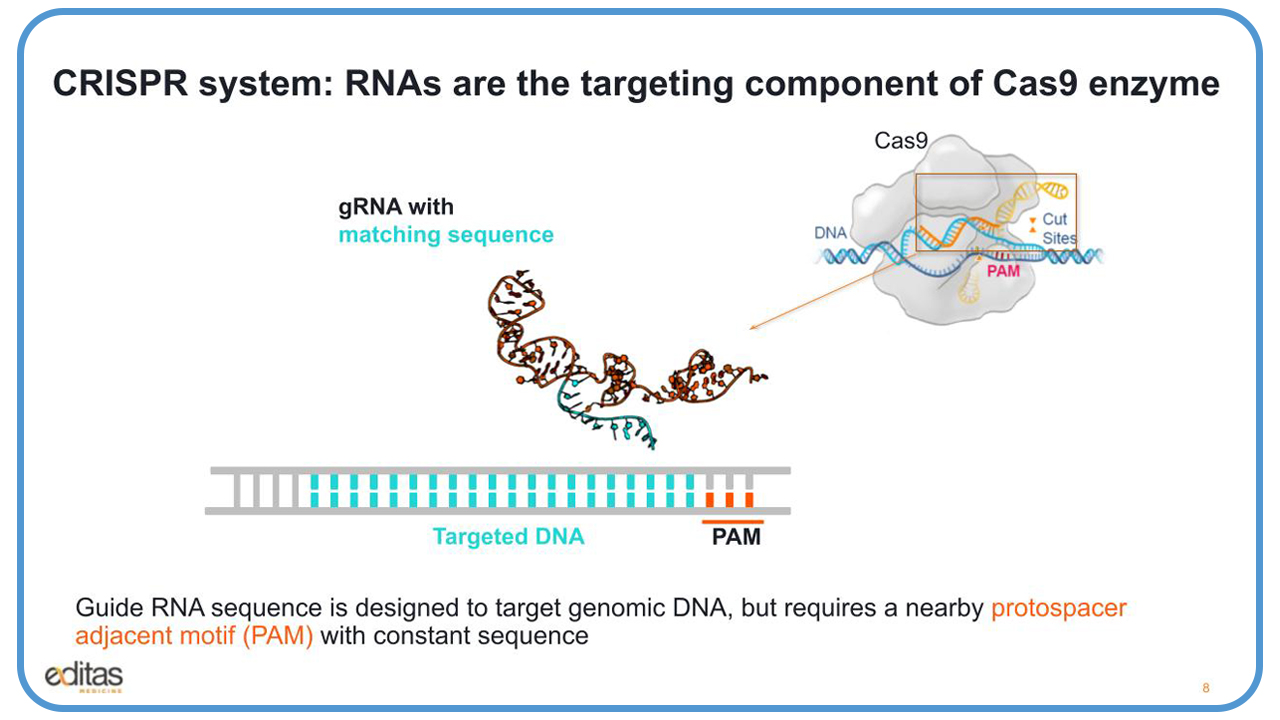
Looking at this slide, you’ll see in the middle our guide RNA, which is composed of two parts. What’s in orange is noted as the tracer, and that part binds to the enzyme. In blue, that is the guide RNA that’s designed to target the genomic DNA, as you can see on the bottom in blue. But it requires a neighboring protospacer adjacent motif, known as the PAM with the constant sequence that the enzyme needs to recognize and cut.
Editas uses two complementary editing platforms, the Cas9 and the Cas12a. The nuclease and guide complex can precisely locate and cut genomic sites. The enzymes can target multiple sites simultaneously, and the enzymes can be engineered to reach more sites and modulate cutting.
The Cas9 recognizes purine-rich PAM, which is located 3′ to the cut site. It’s naturally a two-part RNA, but it can be combined into a single approximately 100 nucleotide RNA, and Cas9 results in a blunt DNA cut. Cas12a recognizes a pyrimidine-rich PAM located 5′ to the cut site. It’s a 40-nucleotide guide RNA, and Cas 12a creates a 5′ staggered cut resulting in a 4- to 5-nucleotide overhang.
Addressing Mutations with CRISPR
CRISPR/Cas9 can address numerous mutations. You could take the enzyme complex to the guide RNA, also known as an RNP, ribonucleoprotein, and you could treat with one RNP to cut over the mutation to create a small insertion, a deletion, or you could take two RNPs and cut on either side of the mutation to delete the mutation. Both result in non-homologous end joining to disrupt a gene or eliminate a disease-causing mutation.
You could take an RNP and cut the genomic DNA, and in the presence of a double stranded DNA, either an AAV or a plasmid, you could insert that piece of DNA into the genome using homology DNA repair. The aim is to promote expression of correct DNA sequences.
Today what I’ll focus on, and show you data for, is the cut-and-remove approach to treat LCA10.
Gene Editing to Repair Retinal Dystrophy
LCA10 stands for Leber Congenital Amaurosis type 104. It is an autosomal recessive condition, which is a result of a biallelic loss of function at chromosome 12 of the CEP290 gene, and it results in severe retinal dystrophy. The main mutation is on intron 26, and it generates a cryptic splice donor site IVS26.
CEP290 is present in the connecting cilium, between the outer and inner segment of the wild-type floater receptors. It’s important for ciliogenesis, ciliary trafficking, and outer segment function and structure.
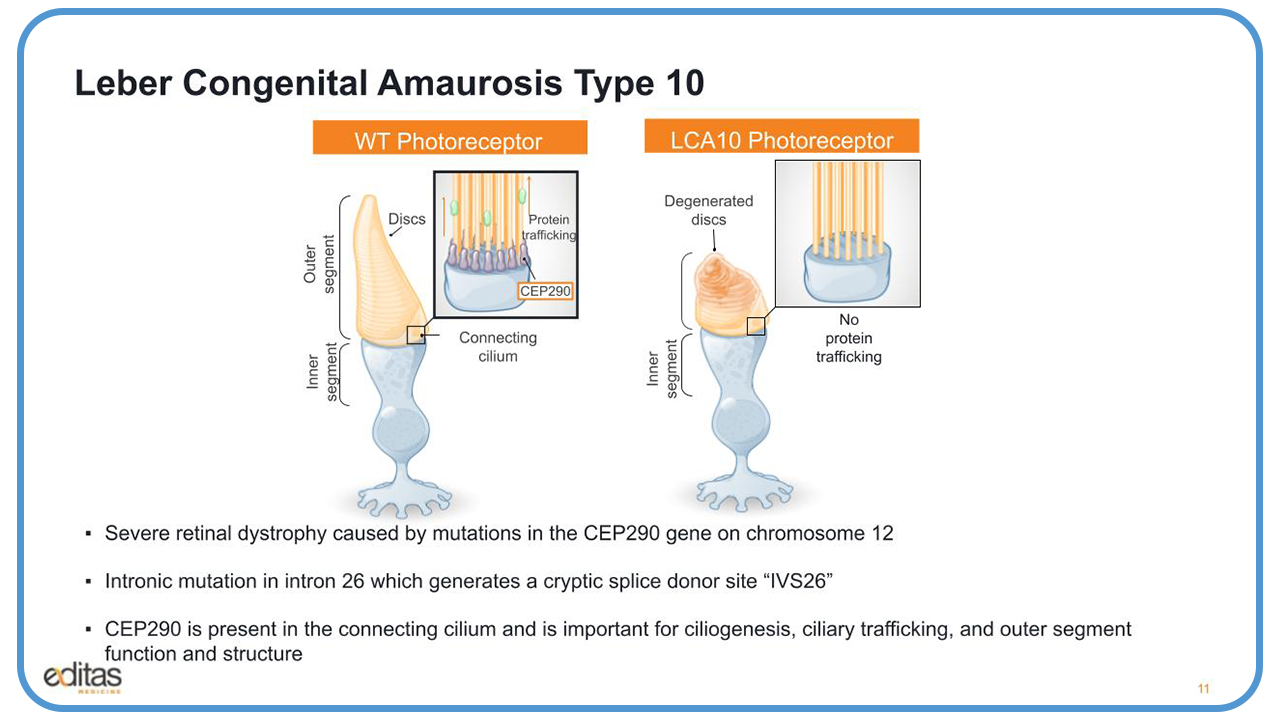
On the right of this slide, we see what happens when there’s mutation to CEP290. There’s no ciliogenesis/ There’s no protein trafficking. And you get the generated discs in the outer segment of the photoreceptor.
The CEP290 transcript is very large. It’s 7.5 kbs, and it’s too large to package an AAV for traditional AAV gene therapy. So, we’re using gene editing to repair CEP290 splicing defect.
As I mentioned, IVS26 is a point mutation which results in a cryptic spliced donor site between Exon 26 and 27. So in the presence of the mutation during transcription, you have 128 bp fragment of the intron that gets inserted between Exon 26 and 27, which results in a premature stop codon and a truncated non-functional protein.
We’re using EDIT-101 to correct this mutation5. EDIT-101 is an AAV that expresses two guides, guide 323 and guide 64 in addition to the staph aureus Cas9 enzyme6. It will either cut and delete the mutation or invert that segment, which will result in the correct splicing and a correct full-length functional CEP290 protein.
Challenges With Multiple Gene Edits
Now there are challenges with PCR-NGS assays when making multiple edits. As I mentioned before, there are two cuts at the CEP290 mutation site, and they’re 1.1 kb apart. So, if you want to set up PCR assays, you’ll need to set up at least three amplicons, one targeting each of the cut sites and then another one for the productive deletion.
And if you want to look at the productive inversion, then you need two additional amplicons. So even with rigorous standards, it’s difficult to cross-compare assays.
With ddPCR, you can sufficiently measure the deletion. There is a probe set in the middle of the 1.1 kb region that is deleted upon editing, and it’s a loss of signal assay. However, if you have an inversion, you won’t be able to distinguish it from the wild-type locus.
The Power of UDiTaS
So, we’ve developed this Uni-Directional Targeted Sequencing methodology, which we call UDiTtaS, and it’s an NGS method for measuring all edited junctions, including the indels.
We have a custom transposon, which is made up of a Tn5 enzyme, and an Illumina adapter that has a unique molecular identifier barcode used to count editing events, along with an i5 pooling barcode to pool a lot of different samples, and the common p5 adaptor sequence to allow for universal amplification with p5 and p7.
The way this works is you take this custom transposon and combine it with genomic DNA extracted from edited cells. And it will fragment approximately 2 kb pieces and tag on the adapters. And then, to enrich these edited fragments, you design a sequence-specific primer upstream or downstream of the target site, and you amplify with the common p5 primer.
In round two, then you use the round two p7 primer, which adds another barcode onto the library. And now you have a full-length sequencing library that you can load onto the Illumina sequencer.
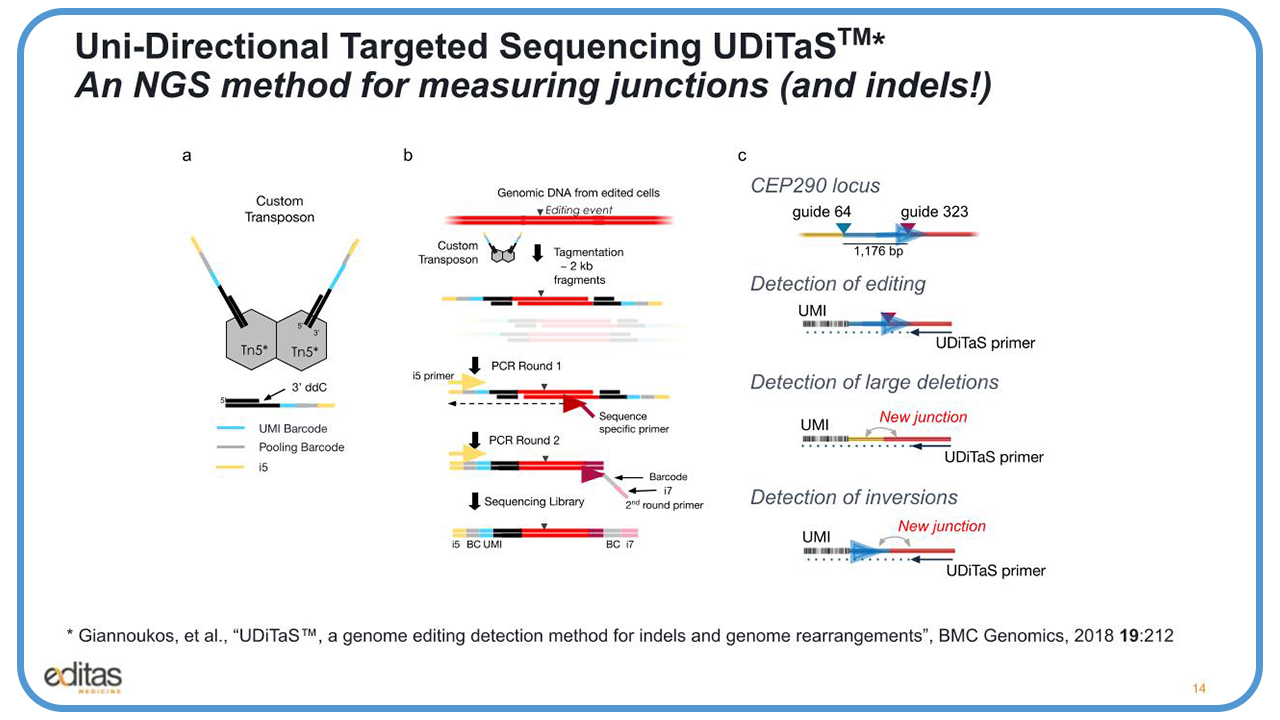
In this slide you see the editing events that we could detect with UDiTaS. And here, if we use one UDiTaS primer, we could look at small indels looking at from the guide 323 site. If you have a large deletion of the 1.2 kb fragment, you’ll create a new junction, and you’ll be able to detect that.
If the 1.2 kb fragment gets removed, inverted, and goes back into the cut site, it’ll create another new junction. And again, we’ll be able to identify that new junction. In addition, we’ll be able to read the UMIs to count the number of edited events.
Proof-of-Concept
We did a proof-of-concept experiment with UDiTaS using U-2 OS cells nucleoffection with plasmids expressing the staph aureus Cas9 in the two guides, 64 and 323, and we were able to see a wide range of edits.
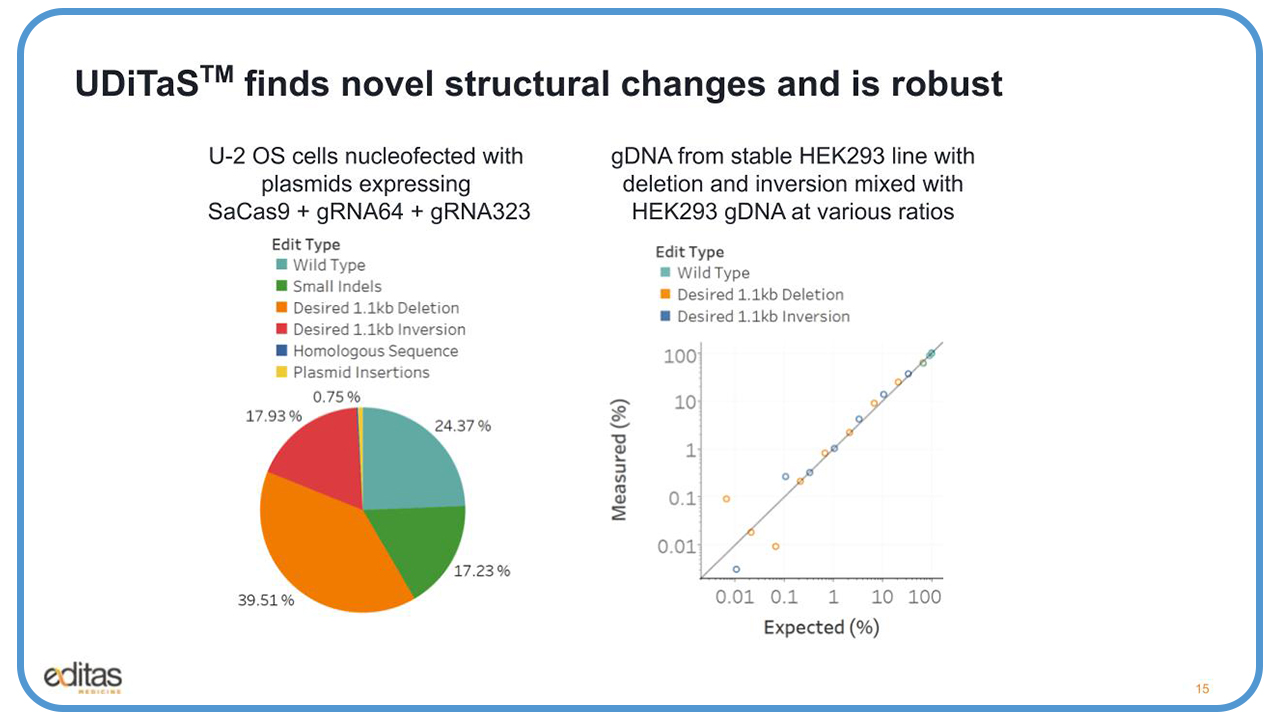
Next, we wanted to see what was the limit of detection of our UDiTaS assay to detect these desired large deletions and inversions. So, we took genomic DNA from an edited stable HEK293 cell line, which had the deletion and inversion, and we mixed it with genomic DNA from the parental HEK293 cells at various ratios. And you could see in this slide a very nice linear detection all the way down to about .1%.
A Not-So-Timid Experiment
We also applied UDiTaS to hundreds of samples from a mouse pharmacology experiment7. We had a humanized CEP290 knock-in mouse model, and we treated the mice with EDIT-101, which is an AAV-5. It consists of the two guides, 323 and 64, and a staph aureus Cas9. The guides are driven by U6 promoter, and the enzyme is the GRK1 promoter.
The AAV was subretinal injected into the eyes of the mice, and we saw a very nice correlation. As the Cas9 mRNA increases, you get an increase in total editing rates using with UDiTaS. We were able to do CEP290 editing in human retinal explants that were treated with EDIT-101.
We did this study with Lions Eye Institute in Florida. They were able to obtain human eyes fixed five hours postmortem. They were able to remove the retina, create three-millimeter punches, and take those punches – with the photo receptors facing down – into a 24-well plate, and then transduce the retina with the AAV.
After four weeks, they were able to collect the genomic DNA, and we were able to process it with UDiTaS to look at the editing. We found that the treated retinal punches with EDIT-101 5e to the 11 viral genomes gave you about approximately 40% editing, and about 15% were functional edits, the inversions and the deletions.
UDiTaS Detects Different Types of Editing Events
UDiTaS can also measure inter-chromosomal translocations. We conducted another experiment with CD4 positive human primary T-cells for nucleoffected with two RNPs. One RNP had a guide to the TRAC locus on chromosome four. The other RNP had a guide to the B2M locus on chromosome 15.
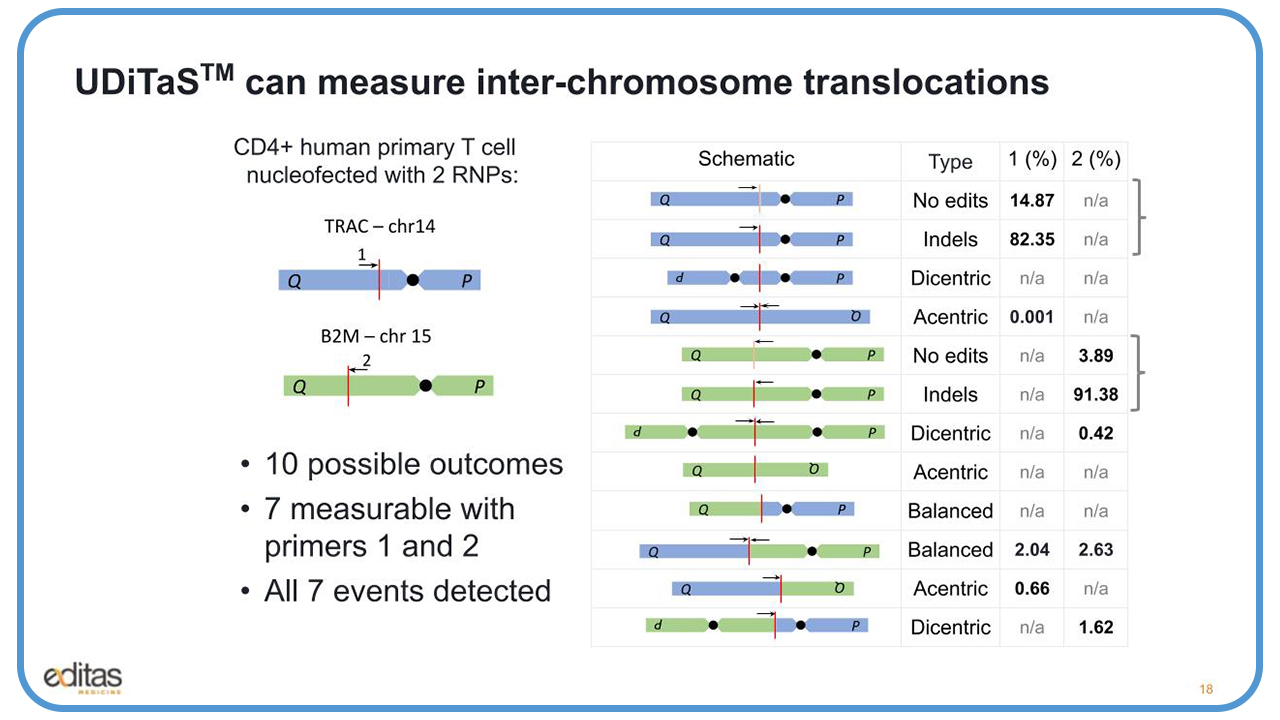
And what we have here, the red line represents the editing event, and the arrows represent the two UDiTaS primers that we used to measure these events. There are 10 possible outcomes, as you see here in the middle under the schematic. First, we have the top in the blue, the events for the TRAC locus. You have the unedited, and below that, the edited. And then the dicentric and acentric translocations, which result from edits and fusion of the sister chromosomes.
Below that in green are the same events that you see at the B2M locus, and below that the translocations between the TRAC and the B2M chromosomes, both the balanced the yield, and the acentric and the dicentric translocations. Seven of these events are measurable by the two primers, primer one and primer two.
When we had very high editing, 82% to 91% for TRAC and B2M, respectively, we also were able to detect with that high editing about 2% to 2.6% balanced translocations. We were also able to create a series of six plasmid standards for the TRAC-B2M translocation, and we added SNPs at every 10 bases so we were able to differentiate between the plasmids. We then diluted these plasmids and serially titrated them from 1.5 million molecules down to 3,000 molecules and spiked them into mouse genomic DNA.
We wanted to determine the sensitivity and the specificity. We ran it through UDiTaS and the Anchored Multiplex PCR, which was developed in John Iafrate’s lab at Massachusetts General Hospital, and is similar to UDiTaS but has additional steps, including fragmentation and adaptalization. We wanted to compare the two methods and found that UDiTaS shows really high accuracy and linearity. We also saw good linearity with the AMP-Seq. However, it is a little bit less accurate, and it’s probably due to the additional steps of fragmentation and adapter ligation and cleanup.
Proof that seqWell Helps
At Editas, we process thousands of samples with UDiTaS every year for all our different programs, and it’s very difficult getting a batch of reagents that are consistent across batches and give us reproducible results. So we reached out to seqWell for help.
The first proof of concept experiment was with the seqWell enzyme in the storage buffer, and we did multiple dilutions to see which one worked the best. We took the best dilutions, made UDiTaS libraries, and sequenced them. The seqWell 1:2 dilution enzyme performed the best. We had the similar recounts, the median size libraries were the same, and we had greater than 85% of the reads aligning to the target.
We were able to see very good reproducibility across batches. For example, we had eight samples that were edited with an AAV that expressed two guides in the Cas12a enzyme, and the goal was to excise the 2.2 kb region.
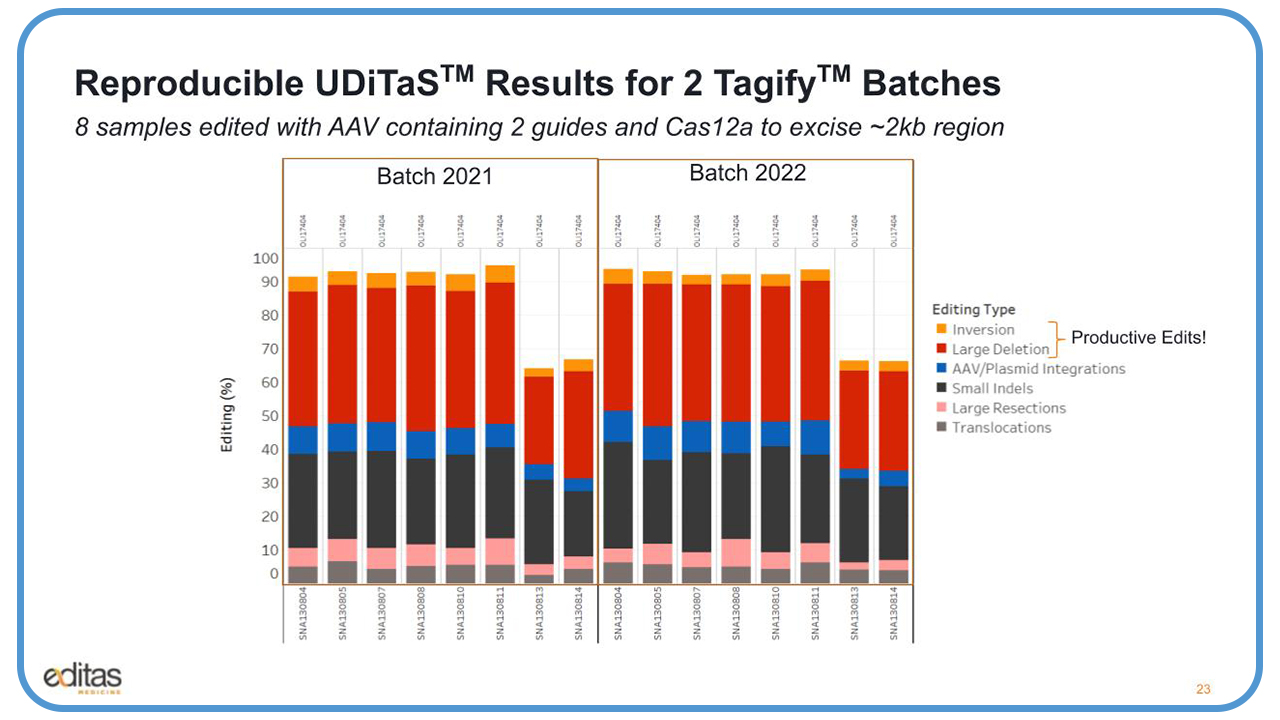
And what you see here on the left with the batch that we had obtained last year, we got really nice editing up to 100%. You can see all the different types of edits, including the productive edits, which are the larger inversions and deletions. We saw some AAV integration, the small indels, and one thing I want to point out in pink are the large resections, which are approximately 50 bases or larger, and in addition to translocations. There is very good consistency across batches, the results look almost identical.
Next, we wanted to try scaling from 96 well reactions to 384 well so we made triplicate reactions in 96 wells and 384 wells. We took an iPSC clone that had been edited two target sites, and we used two UDiTaS primers to look at the results. We saw really nice recounts. The median size fragment size was the same around 325 bp. We saw greater than 95% of the reads mapping to the target region, and we saw greater than 7,000 unique edited events. And what’s clear is that the 384 well reaction captured more unique edited events, and the results are reproducible.
When we looked at these edited events closely, we were able to identify there were two type of events in using each primer. So our primers can detect in this clone that there is a large inversion that has occurred between the two genes.
Closing Remarks
So in summary, UDiTaS can be used to detect all on-target editing events, indels, large deletions, inversions, resections, and inter chromosomal translocations. We’ve used seqWell’s custom Tagify reagent for the UDiTaS process.
We provided seqWell with four adapters. These were complex Illumina, p5-barcoded, barcoded UMI, oligos that were annealed to top and bottom strands. seqWell complexed these adapters with their enzyme and returned ready-to-use reagents. The batches are consistent, and they provide similar tagmentation profiles and editing results. The reactions can be scaled from 96 to 384 wells, and we’ve been able to process thousands of reactions over one year.
A Brief Q&A
How does UDiTaS differ from GUIDE-seq?
Georgia Giannoukos: GUIDE-seq is a discovery assay. It’s a cellular discovery assay of potential off-targets. What usually happens is during a transfection, not only is an RNP introduced into the cells, but so is a double-stranded short oligo. Once editing occurs, the double-stranded oligo gets inserted into the cut site by non-homologous enjoining, and that tags the potential off-target.
You could use UDiTaS to perform the GUIDEseq reaction, and that’s what we do here at Editas. What we do is design two primers targeting the double-stranded oligo. One primer targets the top strand, the other primer targets the bottom strand, and you can look and see where the double-stranded oligo inserts on both sides of the cut. And yes, you could collect those off targets, and then you can verify them either with UDiTaS or another method.
Can you use these Tagify reagents for off-target QC?
Georgia Giannoukos: You can. You can design UDiTaS primers to look at potential off targets. You have to know what the target site would be, and you would design a primer. Not only would you be able to identify the indel, but you would also be able to identify a potential translocation.
With UdiTaS, as we’ve shown in the data today, we could identify potential translocations and in some cases we’ve actually looked at the data more closely. In most of the cases, the translocations are random events due to DNA breaks. However, there are some cases where these are recurring events, and we have been able to identify a translocation with an off-target, and we’ve been able to verify that by going to the off-target location and designing UDiTaS primers from there. So it can be used to assess off-targets, yes.
How many samples can be prepared in a single batch?
Georgia Giannoukos: We could run 96 samples on a plate, and we could run two plates in one day. So, in total, we could prepare 192 samples. We could do more than that. We have four Tagify reagents with four P5 adapters, but we normally run two. And we put those together into a single MiSeq run 2 x 250.
How many reads do you target per sample?
Georgia Giannoukos: We put 50 ng of genomic DNA into the reaction, and that’s about 14,000 genomic target sites. So, when we process the two batches of samples, 192 samples, that comes out to about 50,000 to 100,000 reads, and on average we get about 10,000 unique events for each sample.
How do you minimize the level of background reads from PCR artifacts, and what is the level that you typically see?
Georgia Giannoukos: Well, when we design primers, we usually design three primers per target site and they’re roughly 30mers. We test them, we put them through the UDiTaS process, so we make UDiTaS libraries with unedited genomic DNA. We sequence it, and then we map the reads and choose the best performing primer that gives us greater than 85% of the reads mapping to the target site.
We also size select the libraries before we sequence them between 400 to about 850 bp, and that eliminates a lot of the smaller PCR primers, adapt primer products. So, we don’t see many artifacts because of the size selection as well.
Are there certain gene editing sequences or therapies that are particularly useful for the UDiTaS method versus PCR, for example?
Georgia Giannoukos: So, with UDiTaS I think what is really nice is the fact that you can see if you have any type of translocations, especially if you’re doing multigene editing. You can’t do that with PCR. With PCR, you have to anchor a region with two primers.
What are the current limitations of this assessment?
Georgia Giannoukos: The limitations depend on what the level of editing is. We have been able, as you saw with the data, to identify potential translocations down to a frequency of close to 0.1%, which is the sensitivity of the NGS platform as well.
Could you please describe the main advantages of UDiTaS over other methods?
Georgia Giannoukos: Well, UDiTaS, it’s a very simple assay with relatively low input, 50 ng, and you could get a lot of information from a single reaction. As we showed today, you could assess small indels, large deletions, inversions, translocations. And the other nice thing is that it’s very scalable, and you could process a lot of samples at once.
References
- https://seqwell.com/video/assessing-crispr-on-target-editing-and-structural-changes-with-uditas-using-tagify-reagents/
- https://seqwell.com/expressplex-library-prep-at-agbt-2023/.
- https://pubmed.ncbi.nlm.nih.gov/29562890/
- https://pubmed.ncbi.nlm.nih.gov/30664785/
- https://www.nature.com/articles/s41591-018-0327-9
- https://ir.editasmedicine.com/news-releases/news-release-details/editas-medicine-announces-clinical-data-demonstrating-proof.
- https://www.editasmedicine.com/wp-content/uploads/2019/10/14.pdf