
Why does chronic inflammation lead to cancer? The answer may lie in chromatin.
Numerous publications have linked chronic inflammation to cancer (Biomed Pharmacother, 2023). Even in cases where the patient has recovered from the inflammatory condition, increased cancer risk persists. Understanding the molecular basis of the transition from recovered tissue to tumor could lead to improvements in our ability to diagnose, prevent or treat these cancers.
One of many possibilities for increased cancer risk from recovered cells is epigenetic change. It is well known that cells can retain information of past stress or damage by harboring alterations to the chromatin structure that controls accessibility to DNA. Although gene expression patterns may have returned to pre-inflammation levels, these epigenetic changes in chromatin can persist and offer one theory on how these cells are more prone to becoming cancerous.
Unlocking the biological effects of epigenetic alterations requires tools that can efficiently survey for chromatin changes within cells systematically. One method used to identify and characterize these changes is called SHARE-seq (Single-cell Chromatin Accessibility and mRNA Expression Sequencing).
SHARE-seq is a dual-omics, scalable assay for single-cell chromatin accessibility and mRNA expression profiling.
First reported by S. Ma et al. in Cell, 2020, SHARE-seq simultaneously measures chromatin accessibility and gene expression in individual cells. This dual profiling method offers comprehensive insights into the regulatory landscape and transcriptional states of cells, enabling a deeper understanding of cellular heterogeneity and dynamic biological processes. It combines two methods:
- ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing): This method identifies accessible regions or “footprints” of the genome where regulatory elements like enhancers and promoters are located.
- RNA-seq: This technique profiles gene expression by sequencing RNA molecules, thereby providing information on which genes are actively being transcribed.
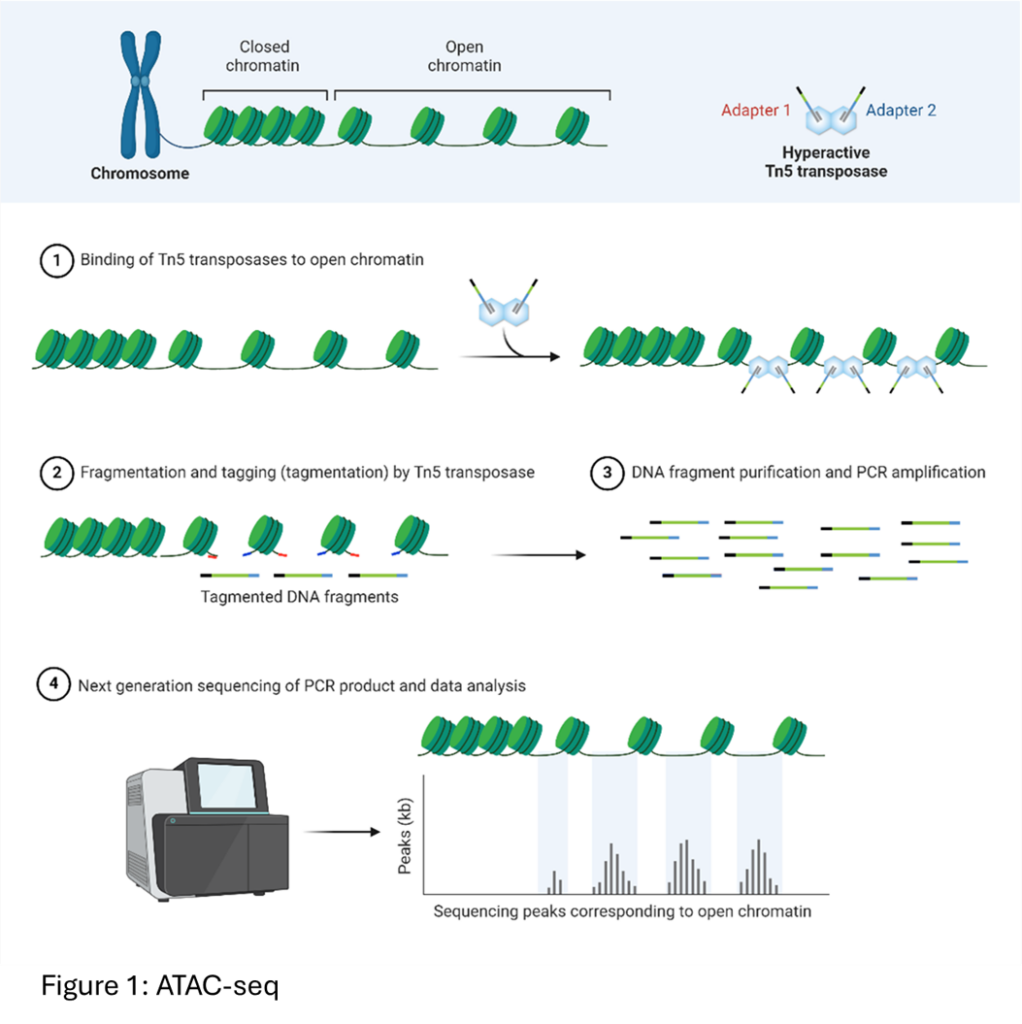
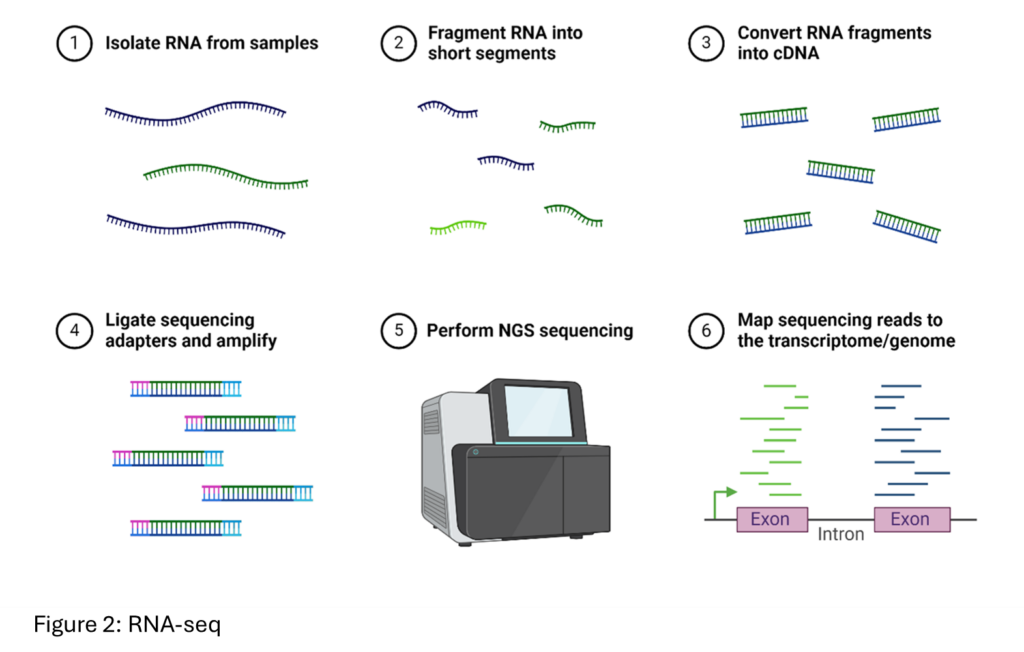
Key features and workflow of SHARE-seq:
- Simultaneous Profiling:
- Chromatin Accessibility: Identifies open chromatin regions using transposase-based methods, similar to ATAC-seq, where the Tn5 transposase integrates sequencing adapters into accessible DNA regions.
- Gene Expression: Measures mRNA levels by capturing and sequencing polyadenylated transcripts, providing a snapshot of the transcriptome.
- Single-Cell Resolution:
- Enables the analysis of chromatin accessibility and gene expression at the single-cell level, offering insights into cellular heterogeneity and dynamic regulatory processes.
- Transposase-Based Library Preparation:
- Uses adapter-loaded transposase to fragment and tag open chromatin regions with sequencing adapters.
- mRNA is captured using oligo(dT) primers that bind to poly-A tails, followed by reverse transcription and second-strand synthesis to create cDNA.
- Sequencing:
- Libraries prepared from both chromatin and mRNA are sequenced using high-throughput sequencing platforms.
- Sequencing data from chromatin accessibility and mRNA expression are mapped to the reference genome and transcriptome, respectively.
- Data Integration:
- Integrates chromatin accessibility and gene expression data to link regulatory elements with their target genes.
Advantages and challenges of SHARE-seq
SHARE-seq provides many advantages – the best of which is that it offers comprehensive profiling of cells at a single cell resolution capturing cellular heterogeneity and dynamic changes that are often masked in bulk assays. In addition, it provides an unbiased view of both the regulatory landscape and transcriptome in single cells, offering a more complete picture of cellular states and functions. SHARE-seq has been shown to be scalable for higher throughput studies of larger numbers of cell populations.
A key technical requirement of assays such as SHARE-seq is the use of a custom transposase-based enzyme with a ligation-enabled payload that permits downstream processing of tagmented cells. Preparation of customized-payload transposase reagents can be a relatively complex task, due to technical variability of published transposase purification and loading protocols.
seqWell Tagify™ custom-loaded transposase reagents provide a commercial source of fully QC’d tagmentation reagents loaded with the necessary adapters that can be used in SHARE-seq and a wide variety of other applications, helping researchers to continue to build new and more reliable assays.
Conclusions
SHARE-seq represents a powerful and versatile tool for studying the interplay between chromatin accessibility and gene expression at single-cell resolution. Its ability to provide a comprehensive view of the regulatory landscape within individual cells opens new avenues for research in cellular biology, development, and disease, offering valuable insights into the underlying mechanisms that govern cellular function and identity.
Watch our webinar to see an example of SHARE-seq in action.
From Colitis to Cancer: Epigenetic Memory View Now
References:
- Contains SHARE-seq protocol: Single-cell multi-scale footprinting reveals the modular organization of DNA regulatory elements. bioRxiv [Preprint]. 2023 Mar 29:2023.03.28.533945. [Version 1] doi: 10.1101/2023.03.28.533945
- Nigam M, Mishra AP, Deb VK, et al. Evaluation of the association of chronic inflammation and cancer: Insights and implications. Biomed Pharmacother. 2023;164:115015. doi:10.1016/j.biopha.2023.115015
- Ma S, Zhang B, LaFave LM, et al. Chromatin Potential Identified by Shared Single-Cell Profiling of RNA and Chromatin. Cell. 2020;183(4):1103-1116.e20. doi:10.1016/j.cell.2020.09.056
- Blog article: A complete guide to understanding ATAC-seq.
Figures created using BioRender.com